
Next-Generation Protein Modeling With Differentiable Retrieval
Why smart retrieval-augmented models are outpacing large protein language models

TL;DR:
• Two paradigms for protein models have emerged: Large protein language models that compress all evolutionary information into parameters vs. retrieval-based models that learn general biochemical constraints while relying on dynamic retrieval of homologous sequences to capture family-specific patterns
• Retrieval-based methods consistently outperform large pLMs on fitness and structure prediction tasks while offering better modularity and adaptability to new sequence data, but are limited by MSA-based retrieval bottlenecks
• End-to-end differentiable retrieval (Protriever) addresses key limitations of alignment-based retrieval and achieves state-of-the-art fitness prediction performance among sequence-based methods with orders of magnitude speedups over current approaches
Two Paradigms for Protein Modeling
Building on our previous discussion about scaling limitations, the protein modeling field has converged on two distinct paradigms for incorporating evolutionary information:
The "Big Model" paradigm: Train massive models that compress all evolutionary patterns and family-specific information directly into their parameters1,2,3. This approach assumes that with enough data and parameters, models can internalize relationships across all protein families. However, scaling protein language models beyond 1-4 billion parameters has shown disappointing returns, suggesting fundamental limitations to this approach.
The "Retrieval-Augmented" paradigm: Train lightweight models that learn general biochemical constraints while dynamically accessing family-specific patterns through retrieval mechanisms4,5,6. Rather than memorizing evolutionary relationships, these models learn how to effectively leverage information from homologous sequences retrieved from external databases.
This retrieval-augmented philosophy aligns with broader trends in machine learning, where hybrid systems combining learned representations with external knowledge sources (eg., access to the internet, proprietary databases) prove increasingly powerful for complex reasoning tasks.
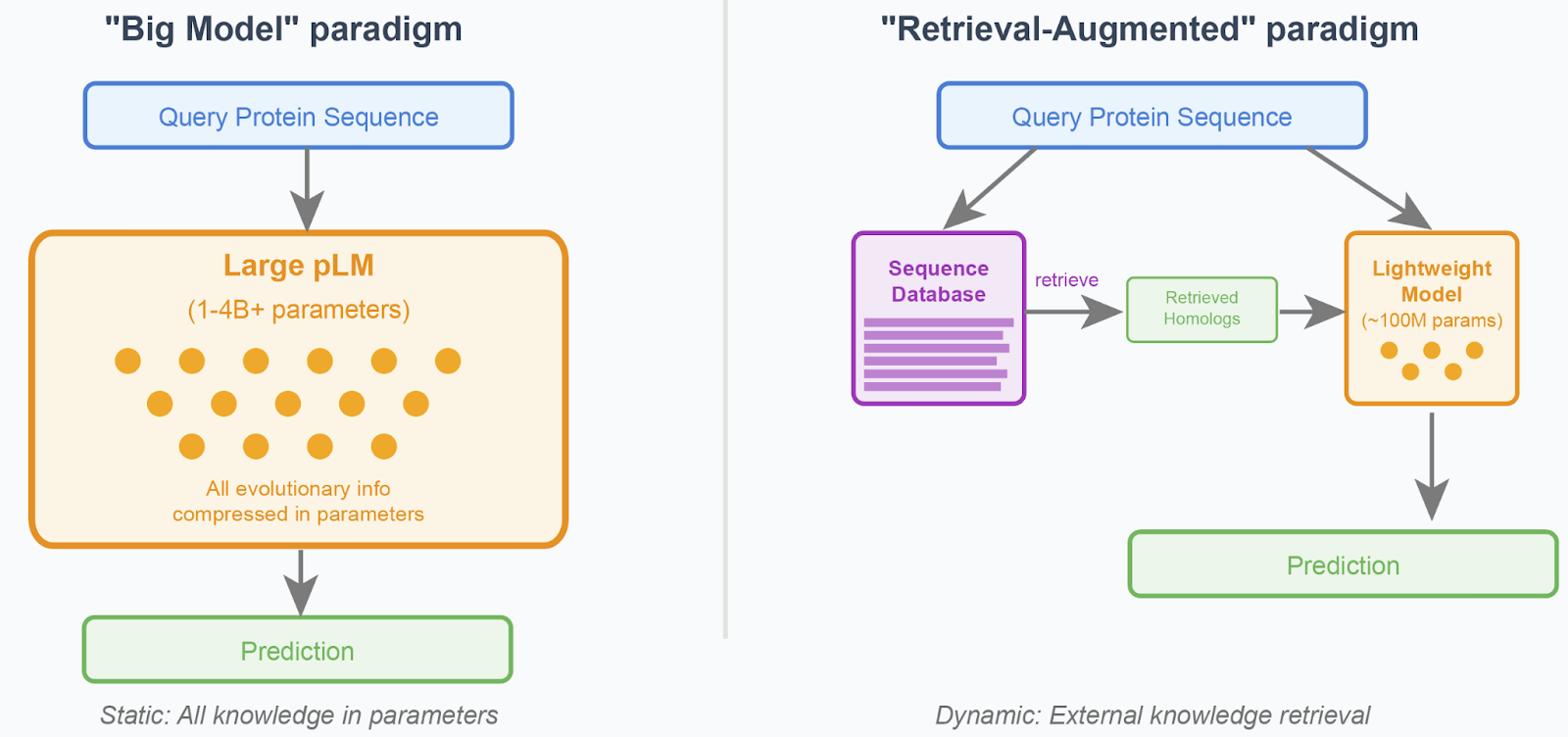
Figure 1: The two core paradigms in protein language modeling. Left: Large pLMs encode all evolutionary information in parameters, requiring massive model capacity. Right: Retrieval-augmented models use lightweight architectures that dynamically access relevant homologous sequences from external databases, enabling more efficient and adaptable systems.
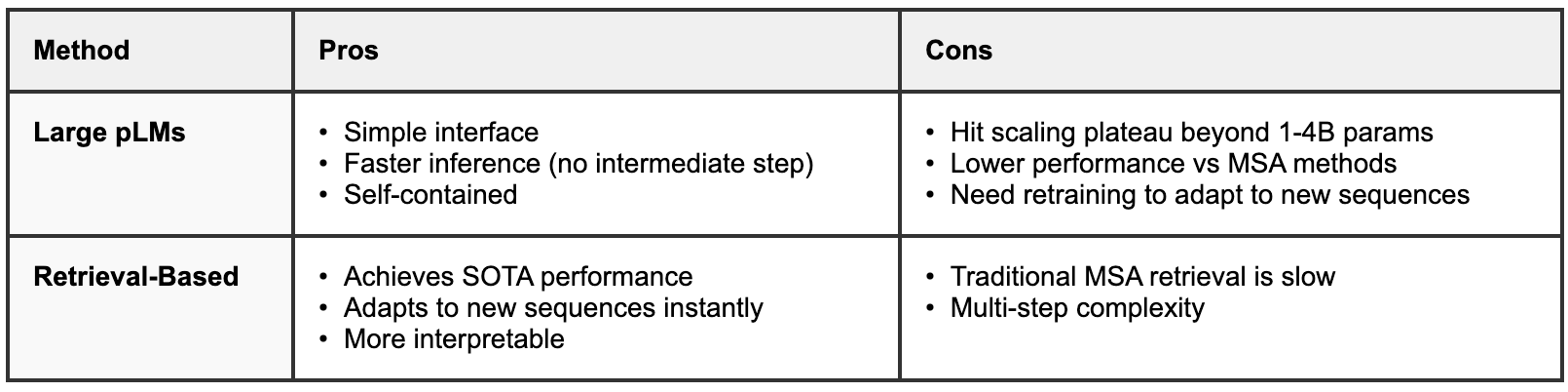
Pros & Cons of Large pLMs vs. Retrieval-Based Methods
The choice between these paradigms involves several trade-offs that have become clearer as both approaches have matured.
Large protein language models offer undeniable advantages in simplicity and deployment. Their straightforward interface—input a sequence, get a prediction—requires no additional infrastructure for homology search or database maintenance. Inference is relatively fast since all knowledge is embedded in the model parameters. However, these benefits come with significant limitations. Beyond the scaling plateau at 1-4 billion parameters, large pLMs face fundamental issues with data quality and representation. Oversized pLMs may actually degrade performance by fitting phylogenetic noise rather than functional constraints7, a problem exacerbated by species oversampling in public databases8,9, which leads models to overfit to taxonomic biases rather than learning generalizable biochemical principles. Additionally, the static nature of these models means they cannot adapt to new sequence discoveries without complete retraining.
Retrieval-based methods consistently achieve state-of-the-art performance on fitness prediction benchmarks precisely because they can focus on learning general biochemical constraints while accessing relevant evolutionary context dynamically. The adaptive nature of these systems means they immediately benefit from new sequence discoveries by simply updating retrieval databases. However, this flexibility comes at the cost of increased complexity, requiring multi-step pipelines and traditionally slow MSA construction methods that can take hours or days for comprehensive homology searches.
Evolution of Retrieval in Protein Modeling
The integration of retrieval within protein modeling has evolved through several generations. First-generation approaches relied purely on protein language models without any retrieval mechanisms, learning evolutionary patterns through self-supervised training on large sequence databases. However, as the field recognized the limitations of single-sequence input approaches, second-generation methods began incorporating retrieval in various forms. Below, we focus on these second-generation approaches that laid the groundwork for modern retrieval-augmented protein modeling.
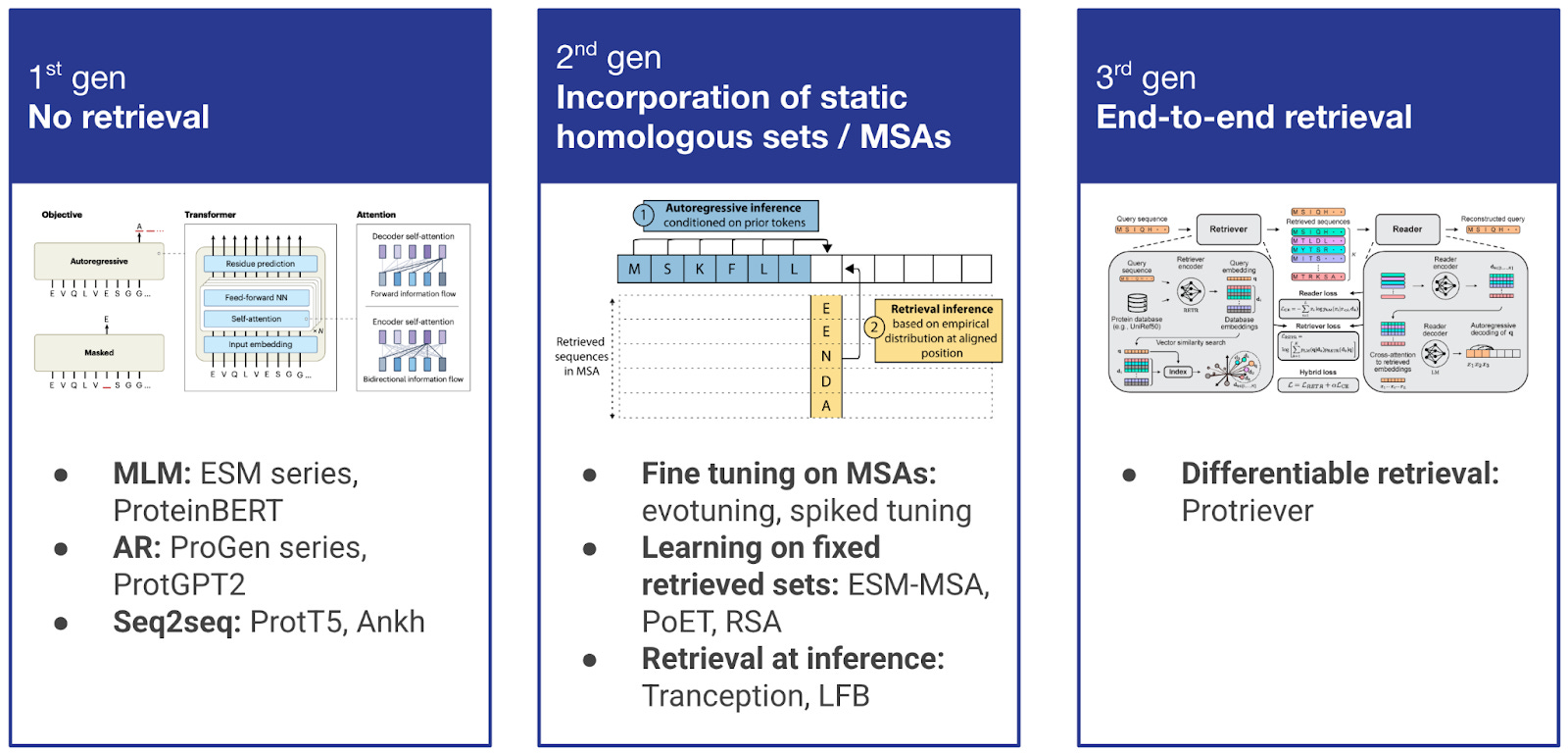
Figure 2. Evolution of retrieval in protein modeling across three generations. 1st generation: Pure protein language models (ESM, ProteinBERT, ProGen, ProtGPT2) with no retrieval mechanisms. 2nd generation: Static incorporation of homologous sequences through fine-tuning on MSAs (evotuning, spiked tuning), learning on fixed retrieved sets (ESM-MSA, PoET, RSA), or retrieval at inference time (Tranception, LFB). 3rd generation: End-to-end differentiable retrieval (Protriever) that jointly optimizes both retrieval and modeling components, enabling dynamic discovery of task-relevant homologs.
Evolutionary Fine-tuning
Early approaches like evotuning extracted MSAs for specific protein families and fine-tuned pretrained pLMs on them. UniRep10 pioneered this approach, though scaling to larger models proved challenging. ESM1v11 addressed this with "spiked fine-tuning," mixing small fractions of family-specific sequences with broader training data to bridge the performance gap with earlier MSA-based methods like DeepSequence.
Learning from fixed retrieved sets
This group of methods first retrieves homologous sets with standard protein sequence retrieval mechanisms, and then learns general distributions across these retrieved sets. Notable examples include:
MSA Transformer4 treats MSAs as 2D objects, using axial attention from computer vision to learn distributions over MSA inputs while keeping computation tractable
PoET6 takes a different approach, linearizing homologous sequences by concatenating them and training a custom decoder transformer to learn distributions of evolutionarily-related sequence sets
Methods like RSA12 retrieve homologs using similarity in embedding space with a frozen transformer encoder, achieving computational speedups but still relying on task-independent retrieval that remains decoupled from the downstream modeling objective.
Retrieval at Inference
Rather than modifying model architectures or training procedures, a different strategy performs retrieval at inference time, offering both performance improvements and flexibility in how evolutionary context is incorporated:
Tranception5/TranceptEVE13 ground pLMs by retrieving MSAs for target sequences, obtaining prior distributions for amino acid frequencies at each position, and combining these with autoregressive predictions from decoder-style transformers
LFB14 (Likelihood-Fitness Bridging) retrieves MSAs and averages mutation effects across multiple forward passes of the pLM with different background sequences to alleviate the overfitting/bias issues discussed above
While these approaches show promise, they rely on traditional alignment-based retrieval that is slow and operates independently of the downstream task.
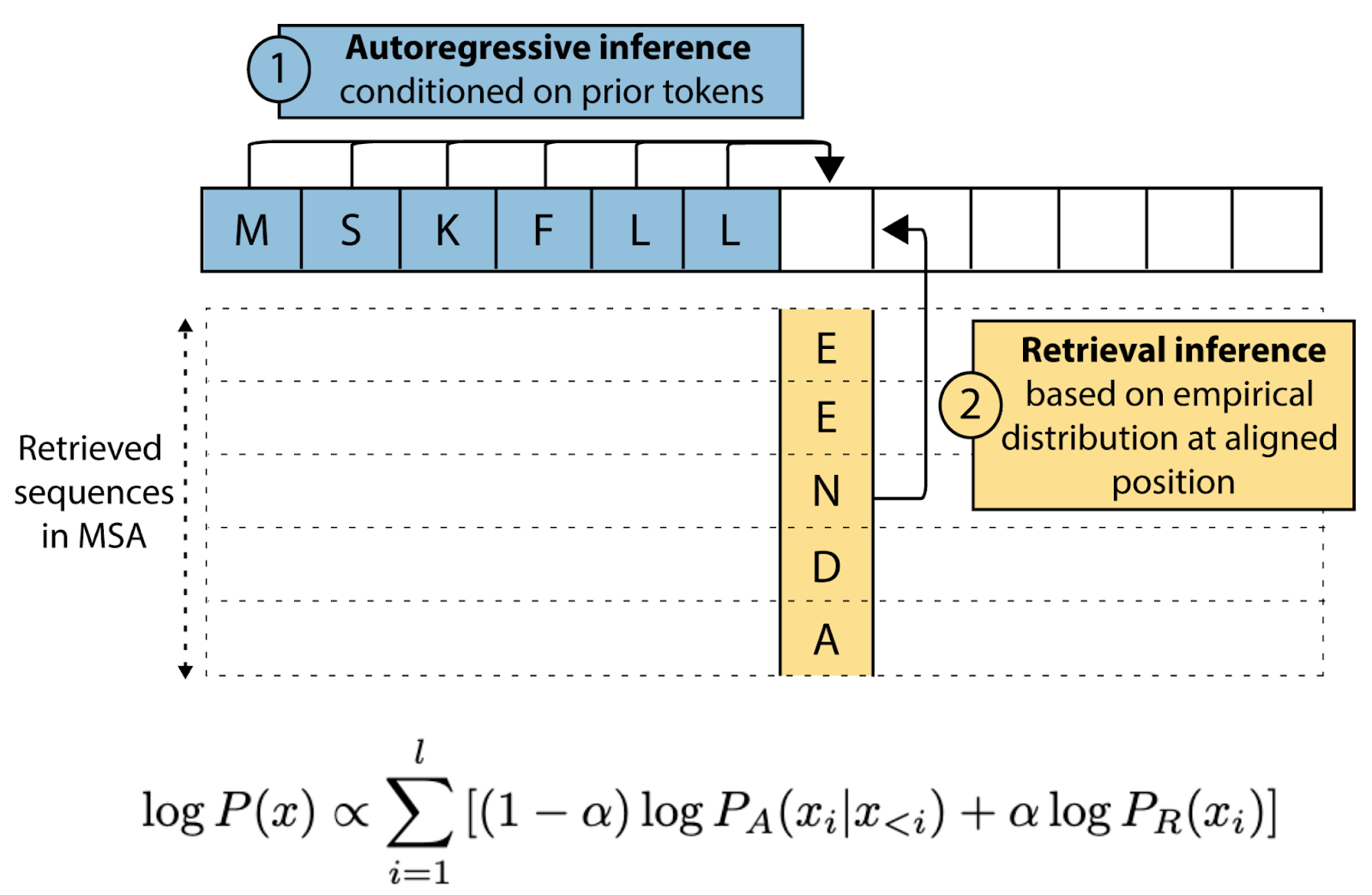
Figure 3: Retrieval at inference with Tranception. The model combines autoregressive predictions from a protein language model with evolutionary priors extracted from retrieved MSAs. At each position, amino acid frequencies from the MSA provide evolutionary context that is integrated with the transformer's learned representations to produce final fitness predictions.
Limitations of Traditional Alignment-Based Retrieval

Figure 4: Traditional two-step protein modeling workflow. Step 1: Retrieve homologous sequences using alignment-based methods (e.g., BLAST, JackHMMER) to construct MSAs. Step 2: Train family-specific models (e.g., PSSM, Potts models, VAE) on the retrieved sequence set. This approach suffers from retrieval being independent of the downstream modeling objective.
Current MSA-based retrieval methods face several fundamental limitations:
Missing distant relationships: MSA-based retrieval often misses distantly related sequences that fall below alignment significance thresholds, and struggles particularly with intrinsically disordered regions15
Structural complexity: Sequences with large insertions, deletions, or structural rearrangements remain difficult to align reliably, despite potential functional relatedness
Task independence: Retrieval operates independently of downstream objectives, relying on alignment heuristics rather than data-driven approaches to identify the most informative homologs
Computational demands: Traditional alignment methods are computationally expensive and poorly suited for large-scale applications like proteome-wide predictions
However, we can overcome these limitations by adapting approaches from the NLP literature, particularly fast vector search systems that power modern RAG architectures16.
Introducing Protriever: End-to-End Differentiable Retrieval
Building on advances in retrieval-augmented generation, we developed Protriever17 - an end-to-end differentiable framework that learns to retrieve relevant homologs and simultaneously trains a sequence model conditioned on these retrieved sequences.

Figure 5: Protriever framework architecture. The system consists of three main components: (1) a learned retriever that embeds sequences and performs vector similarity search, (2) a fast vector index of protein sequences, and (3) a reader model that performs sequence reconstruction conditioned on retrieved homologs. During training, gradient feedback from the reader teaches the retriever which sequences are most useful for the downstream task.
Architecture Overview
Protriever consists of three main components:
Retriever: A transformer encoder (initialized with ESM-218 weights) that learns to embed protein sequences in a space where homologous proteins have high similarity scores
Vector Index: A fast, scalable index of ~62 million UniRef50 protein embeddings using Faiss19 for efficient similarity search
Reader: A sequence modeling architecture (we use PoET6) that performs the target task conditioning on retrieved homologous sequences
Joint Training Strategy
The key innovation is joint optimization: during training, the reader learns which retrieved sequences provide useful context for sequence reconstruction, providing gradient feedback to the retriever. This allows the retriever to learn representations optimized for the specific downstream task rather than generic sequence similarity.
The training process uses a hybrid loss function that combines both retrieval and reader objectives. The reader loss applies standard autoregressive language modeling to reconstruct the query sequence given the retrieved homologs. The retriever loss uses the EMDR20 (End-to-end training of Multi-Document Reader and Retriever) approach, which treats retrieved sequences as latent variables and optimizes the retriever to find sequences that actually help the reader reconstruct the query. The final training objective balances these two components, ensuring that the retriever learns to find sequences that are actually useful for the reader's sequence modeling task.
Performance Results
State-of-the-art accuracy: Protriever achieves the highest performance among sequence-based models on ProteinGym21 benchmarks:
Spearman correlation: 0.479 (vs. 0.470 for previous best PoET)
Performance across metrics: Best results on AUC (0.762), MCC (0.374), NDCG (0.788), and top-K recall (0.229)
Massive retrieval speed improvements: Two orders of magnitude faster than existing retrieval approaches, including the recently introduced MMseq2-GPU22:
Protriever: ~0.005 seconds per query
MMseqs2-GPU: ~0.6 seconds per query
MMseqs2: ~16.9 seconds per query
JackHMMER: ~2500 seconds per query
Consistent performance: Strong results across different protein families and evolutionary contexts, with particularly notable improvements on prokaryotes and viruses.

Figure 6: Protriever performance on ProteinGym benchmark. Comparison of Protriever against baseline methods across all key evaluation metrics (Spearman correlation, AUC, MCC, NDCG, and top recall), demonstrating state-of-the-art performance among sequence-based models.

Figure 7: Retrieval speed comparison across different query batch sizes. Protriever's vector-based search achieves consistent sub-millisecond retrieval times per query, demonstrating 100x speedup over MMseqs2-GPU and 1000x speedup over traditional MMseqs2, while maintaining scalability as the number of queries increases.
Key Advantages
SOTA performance: Achieves the best results on protein fitness prediction among sequence-based methods
Architecture agnostic: Framework can adapt to different retriever and reader architectures, offering flexibility for future improvements
Task agnostic: The same architecture can be repurposed to tackle other downstream tasks such as tertiary structure or function prediction
Significant speedups: Vector-based search enables real-time applications and proteome-scale analyses
Adaptive databases: Can immediately leverage new sequences by updating the index without model retraining. The approach also supports domain-specific specialization - the same trained retriever can work with focused databases like GISAID23 for viral sequences or proprietary datasets for industrial applications
Interpretability: Visibility into the particular sequences selected by the retriever to augment downstream task predictions
Conclusion
The protein modeling field is experiencing a paradigm shift toward retrieval-augmented approaches that combine the best of both worlds: the generalization capabilities of large language models and the evolutionary specificity of family-based methods.
Protriever demonstrates that:
End-to-end differentiable retrieval can match or exceed the performance of traditional MSA-based methods
Vector search can replace computationally expensive alignment algorithms without sacrificing accuracy
Joint training allows models to learn task-specific notions of sequence similarity
Retrieval-based architectures offer superior adaptability and maintainability compared to approaches purely based on scaling pretraining
As the field continues to evolve, we expect retrieval-augmented protein models to become the dominant paradigm, offering the performance needed for critical applications while maintaining the flexibility to adapt to rapidly expanding sequence databases and evolving scientific understanding.
The future of protein modeling lies not in building ever-larger models that attempt to memorize all of biology, but in developing intelligent systems that can dynamically access and reason over the vast evolutionary record encoded in sequence databases.
For more details on the technical implementation and comprehensive benchmarks, see our full paper.
Thanks
Special thanks to Ruben Weitzman and Peter Mørch Groth for their feedback on this blog post, and for the rest of the Marks lab for helpful discussions.
References
Rives et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. PNAS (2021). https://www.pnas.org/doi/full/10.1073/pnas.2016239118
Madani et al. Large language models generate functional protein sequences across diverse families. Nature Biotechnology (2023). https://www.nature.com/articles/s41587-022-01618-2
Chen et al. xTrimoPGLM: unified 100-billion-parameter pretrained transformer for deciphering the language of proteins. Nature Methods (2025). https://www.nature.com/articles/s41592-025-02636-z
Rao et al. MSA Transformer. ICML (2021). https://proceedings.mlr.press/v139/rao21a.html
Notin et al. Tranception: Protein Fitness Prediction with Autoregressive Transformers and Inference-time Retrieval. ICML (2022). https://proceedings.mlr.press/v162/notin22a.html
Truong Jr et al. PoET: A generative model of protein families as sequences-of-sequences. NeurIPS (2023). https://proceedings.neurips.cc/paper_files/paper/2023/hash/f4366126eba252699b280e8f93c0ab2f-Abstract-Conference.html
Weinstein et al. Non-identifiability and the Blessings of Misspecification in Models of Molecular Fitness. bioRxiv (2022). https://doi.org/10.1101/2022.01.29.478324
Hopf et al. Mutation effects predicted from sequence co-variation. Nature Biotechnology (2017). https://www.nature.com/articles/nbt.3769
Ding et al. Protein language models are biased by unequal sequence sampling across the tree of life. bioRxiv (2024). https://www.biorxiv.org/content/10.1101/2024.03.07.584001v1
Alley et al. Unified rational protein engineering with sequence-based deep representation learning. Nature Methods (2019). https://www.nature.com/articles/s41592-019-0598-1
Meier et al. Language models enable zero-shot prediction of the effects of mutations on protein function. NeurIPS (2021). https://proceedings.neurips.cc/paper/2021/hash/f51338d736f95dd42427296047067694-Abstract.html
Ma et al. Retrieved Sequence Augmentation for Protein Representation Learning. EMNLP (2024). https://aclanthology.org/2024.emnlp-main.104/
Notin et al. TranceptEVE: Combining Family-specific and Family-agnostic Models of Protein Sequences for Improved Fitness Prediction. bioRxiv (2022). https://www.biorxiv.org/content/10.1101/2022.12.07.519495v1
Pugh et al. From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models. bioRxiv (2024). https://www.biorxiv.org/content/10.1101/2024.05.20.655154v1
Riley et al. The difficulty of aligning intrinsically disordered protein sequences as assessed by conservation and phylogeny. PLOS ONE (2023). https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0288388
Lewis et al. Retrieval-Augmented Generation for Knowledge-Intensive NLP Tasks. NeurIPS (2020). https://proceedings.neurips.cc/paper/2020/hash/6b493230205f780e1bc26945df7481e5-Abstract.html
Weitzman et al. Protriever: End-to-End Differentiable Protein Homology Search for Fitness Prediction. arXiv (2024). https://arxiv.org/abs/2506.08954
Lin et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science (2023). https://www.science.org/doi/10.1126/science.ade2574
Faiss: A library for efficient similarity search. https://ai.meta.com/tools/faiss/
Sachan et al. End-to-End Training of Neural Retrievers for Open-Domain Question Answering. ACL (2021) https://aclanthology.org/2021.acl-long.519/
Notin et al. ProteinGym: Large-Scale Benchmarks for Protein Design and Fitness Prediction. NeurIPS (2023). https://papers.nips.cc/paper_files/paper/2023/hash/cac723e5ff29f65e3fcbb0739ae91bee-Abstract-Datasets_and_Benchmarks.html
Kallenborn et al. GPU-accelerated homology search with MMseqs2. bioRxiv (2024). https://www.biorxiv.org/content/10.1101/2024.11.13.623350v6
GISAID: Global Initiative on Sharing Avian Influenza Data. https://gisaid.org/