
Have We Hit the Scaling Wall for Protein Language Models?
Beyond Scaling: What Truly Works in Protein Fitness Prediction

Disclaimer
This is not a Gary Marcus-style post for protein language models. I personally hope the answer is no, and there are a few emerging ideas (see end of post) with the potential to overcome current limitations. But, for now, naively scaling pLMs has led to underwhelming performance on the tasks that matter.
TLDR
We just released ProteinGym v1.3, now featuring over 90 baseline models (including recent models Progen3, ESM3/C, and xtrimoPGLM)
The new leaderboard tells a clear story: multimodal models that combine Multiple Sequence Alignments (MSAs) and structure outperform others for zero-shot fitness predictions. Both modalities are useful in different settings
Even very simple methods leveraging these two modalities significantly outperform billion-parameter sequence models
Scaling protein language models (pLMs) does not seem to help beyond 1-4B parameters
We conclude with some thoughts on what the field should focus on going forward to continue driving progress
Introduction
The protein sequence-function relationship represents a fundamental challenge in computational biology, with profound implications for drug discovery, protein engineering, and our understanding of genetic disease mechanisms. Significant progress is being made along two complementary axes. On the data front, the emergence of megascale assays, such as the ones from the Rocklin lab1 or Lehner lab2, combined with initiatives like the Open Datasets program3 from the Align Bio foundation are accelerating the release of high-throughput experimental data. Simultaneously, modeling innovations are pushing the boundaries of what's possible with computational approaches. The parallel between sequences of characters in natural language and sequences of amino acids in proteins4, coupled with the undeniable successes of scaling in LLMs5, had led many (including myself) to believe scaling protein Language Models (pLMs) would drive the field forward. So, a few years later after the emergence of these ideas, where are we?
ProteinGym, One Year Later
We released ProteinGym6 at NeurIPS at the end of 2023. ProteinGym is a comprehensive suite benchmark suite for protein fitness prediction and design that comprises over 250 curated deep mutation scanning (DMS) assays (~3M mutated sequences), and a clinical benchmark w/ annotations of human mutations by domain experts. We implemented a broad collection of model baselines and systematically evaluated them in both zero-shot and few-shot settings.
This blog focuses specifically on DMS benchmarks in the zero-shot setting. Last week, we released version 1.3, which now includes over 90 baselines (compared to ~50 in v1.0). The field has advanced rapidly over the past 12 months, so it felt useful to look back and reflect on what appears to be the most effective modeling strategies.
But before diving deeper, a quick background if you are new to this field:
What are deep mutational scanning (DMS) assays? DMS assays7 are high throughput experimental assays in which various mutations (single or multiple substitutions, short insertions or deletions) are applied to a reference sequence (usually a reference protein sequence found in nature) and measure the effects of these mutations on a property of interest of that protein (eg., thermostability, catalytic activity, ability to bind to the right target).
Why does performing well on ProteinGym matter?
Clinical relevance: A significant proportion of human genetic variation in coding regions involves small numbers of mutations relative to wild-type. Accurately predicting these effects has immediate implications for personalized medicine, in particular without relying on existing labels8
Iterative design: In ML-driven directed evolution campaigns, predicting mutation effects in the neighborhood of a query sequence is essential for navigating the fitness landscape effectively.
Biochemical understanding: To perform well on these benchmarks, models have to capture a nuanced understanding of the biochemical constraints for the corresponding proteins as they have to be able to detect subtle effects resulting from minor sequence changes.
Shallow to deep generalization: A model that cannot judge the effect of a single point mutation is unlikely to score (let alone generate) long multi‑mutant variants correctly.
A model that cannot judge the effect of a single point mutation is unlikely to score (let alone generate) long multi-mutant variants correctly.
The Rise of Multimodal Models of Structure and MSAs
We focus here on 20 of the most popular and/or best-performing protein model architectures from the latest ProteinGym benchmarks. For a given model architecture, we focus on the best-performing checkpoint when multiple versions exist (e.g., ESM2 650M is the top performer in the ESM2 series).

Figure 1: ProteinGym leaderboard. Performance comparison of 20 leading protein models on ProteinGym v1.3, showing average Spearman correlation (left) and NDCG at 10% quantile (right) across 217 DMS assays.
Figure 1 shows the average Spearman correlation (left) and NDCG at 10% quantile (right) across 217 assays from the zero-shot DMS substitutions benchmark. These metrics offer complementary perspectives: Spearman assesses a model's ability to correctly rank mutations from most deleterious to most beneficial (ideal for overall mutation effect prediction), while NDCG measures how well a model ranks proteins by experimental fitness values, giving greater weight to accurately positioning beneficial mutations at the top (crucial for protein design applications).
Several clear trends emerge:
MSAs + Structure: Models accepting only single sequences as input (e.g., ESM, Progen, xtrimoPGLM) are consistently outperformed by those leveraging either structure or MSAs — with the best models (e.g., VenusREM, S3F-MSA) incorporating both.
Task-specific strengths: The top-performing models for Spearman correlation (mutation effect prediction focus) aren't necessarily the best based on NDCG (design focus). Generally, structural information provides an edge for the former, while MSAs appear critical for the latter.
Limits of scaling: Model size has surprisingly little impact on performance beyond a certain point (more on this below), with some of the best-performing approaches being relatively compact architectures.
Rapid progress: The field is advancing at an extraordinary pace—SaProt, which topped the leaderboard when we released v1.1 in mid-2024, has now fallen outside the top 10 on both metrics.
Performance Breakdown by Function Type and Taxon
While the overall leaderboard reveals high-level modeling strategies with the greatest impact, one core motivation behind building ProteinGym was to collect enough assays to enable more granular analyses, for example based on taxonomic origin, function type, MSA depth, and mutational depth. For brevity, I'll focus on the first two dimensions below (function type and taxonomy) but, if you are curious, definitely check out the other ones on the website or the paper.

Figure 2: Performance by function type. Model performance varies significantly depending on the protein function being assayed. Structure-based models excel at stability prediction, while MSA-based approaches better capture catalytic activity and organismal fitness.
We categorize assays into five groups based on the function being measured (e.g., stability, catalytic activity, expression). The results reveal:
Structure-dependent functions: Structural information is particularly valuable for stability and binding prediction, as one might expect. For more on this topic, read this paper9.
MSA-dependent functions: MSAs provide crucial information for predicting organismal fitness and catalytic activity.
Note on the stability deep dive: Models leveraging MSAs appear to have relatively less compelling performance on stability assays. This may stem from a fundamental mismatch between what these models predict and what stability assays actually measure. When mutations affect evolutionarily conserved positions (such as catalytic sites), they might have catastrophic effects on overall protein function while showing only minimal impact on stability in isolation. MSA-based models correctly identify these mutations as functionally deleterious based on evolutionary conservation patterns but are then penalized in stability benchmarks where these functional constraints aren't captured by the experimental readout (h/t Nathan Rollins for pointing this out). This observation should serve as a reminder that experimental assays have their own limitations and biases, and shouldn't be treated as perfect ground truth when evaluating computational models.

Figure 3: Performance by taxon. Most models show a pronounced performance gap between viral and non-viral proteins, with ESM-based models particularly affected despite their strong performance on eukaryotic and prokaryotic proteins.
When we partition assays based on the taxonomic origin of their reference sequences, a striking pattern emerges between viral and non-viral proteins (eukaryotes & prokaryotes). Setting aside models like ESM3-open that specifically excluded viral proteins from training data, the most notable observation is the substantial performance drop on viral proteins from the ESM series (ESM2 and ESM-C shown here; but we had observed similar patterns with ESM1v in the Tranception paper10).
This limitation propagates to models like ProSST (and thus VenusREM), S3F, and ProtSSN, which rely on ESM2 embeddings for initial residue representation. While ESM models generally perform excellently, researchers should exercise caution when applying them to viral proteins, especially in zero-shot settings, as performance is likely to be suboptimal. For an excellent deep dive into this question, check out this paper11: https://openreview.net/forum?id=DvC6VL7TJK.
The Scaling Wall in Protein Fitness Prediction
A puzzling observation from Figure 1: protein language models, even those exceeding 10 billion parameters, are nowhere near the top of these rankings. Why is that?

Figure 4: Fitness prediction performance vs model scale. The Zero-shot fitness prediction performance for protein language models of various sizes, showing initial gains that plateau around 1-4B parameters before declining at larger scales. A bit of retrieval provides a clear performance boost in Tranception10.
In spring 2022, together with colleagues at LightOn, we published the first analysis on the scaling laws for protein language models12. At that time we had scaled pLMs up to 1.2B parameters, and up to that point scaling kept leading to higher performance on downstream tasks. However, subsequent works showed that, beyond that point, further scaling gains did not materialize. The plot above shows zero-shot fitness prediction performance across multiple pLM architectures implemented in ProteinGym. From 1B to ~4B, performance plateaus with minimal gains. Beyond 5B parameters, performance consistently declines with more scaling.
Sergey Ovchinnikov has an insightful back-of-the-envelope calculation (see Zhang et al13) estimating the parameter count needed if we assume protein language models primarily learn evolutionary couplings—interestingly, this calculation yields approximately 4B parameters, aligning with our empirical observations. In a separate work, Weinstein et al14 had previously presented compelling arguments for why oversized pLMs might actually degrade performance by fitting phylogenetic noise rather than functional constraints.
Contrary to expectations from other domains, scaling protein language models shows diminishing returns beyond 1B parameters and actual performance degradation beyond 4B—suggesting we may have hit a scaling wall for pLMs trained on evolutionary sequences.
Would more data help? Expanding to substantially larger sequence datasets (e.g., from metagenomic databases) might mitigate the performance collapse by directing model capacity toward fitting new protein families. However, this would likely only improve performance on our benchmarks if that leads to an increase in relevant diversity—that is, novel sequences within the same protein families or from the same underlying domains as the ones from ProteinGym.
What About Genetic Language Models?
Over the past 3 years, in parallel to the development of protein language models, significant progress has been made in training genetic Language Models (gLMs), ie. models learning from large repertoires of DNA sequences, such as the Evo series. Evo215 is a 40B-parameter model trained on 9.3T nucleotide tokens from long genomic sequences across species. It represents an impressive computational achievement with innovative approaches for probing the biological knowledge learned by the model. Scoring this model on ProteinGym requires mapping protein sequences back to the corresponding DNA sequences -- this is a bit of a tedious process that we will eventually extend to the full benchmark. For now, we've completed this mapping for a representative subset of 22 assays, balanced across function types and taxa. To ensure fair comparison, this subset includes 11 prokaryotic and 11 eukaryotic assays but no viral assays, since viral genomes were excluded from Evo2's training data -- as noted in the Evo2 paper, the performance of the model on viral assays is close to random, and we observe the same on a subset of 4 viral assays on our end as well.
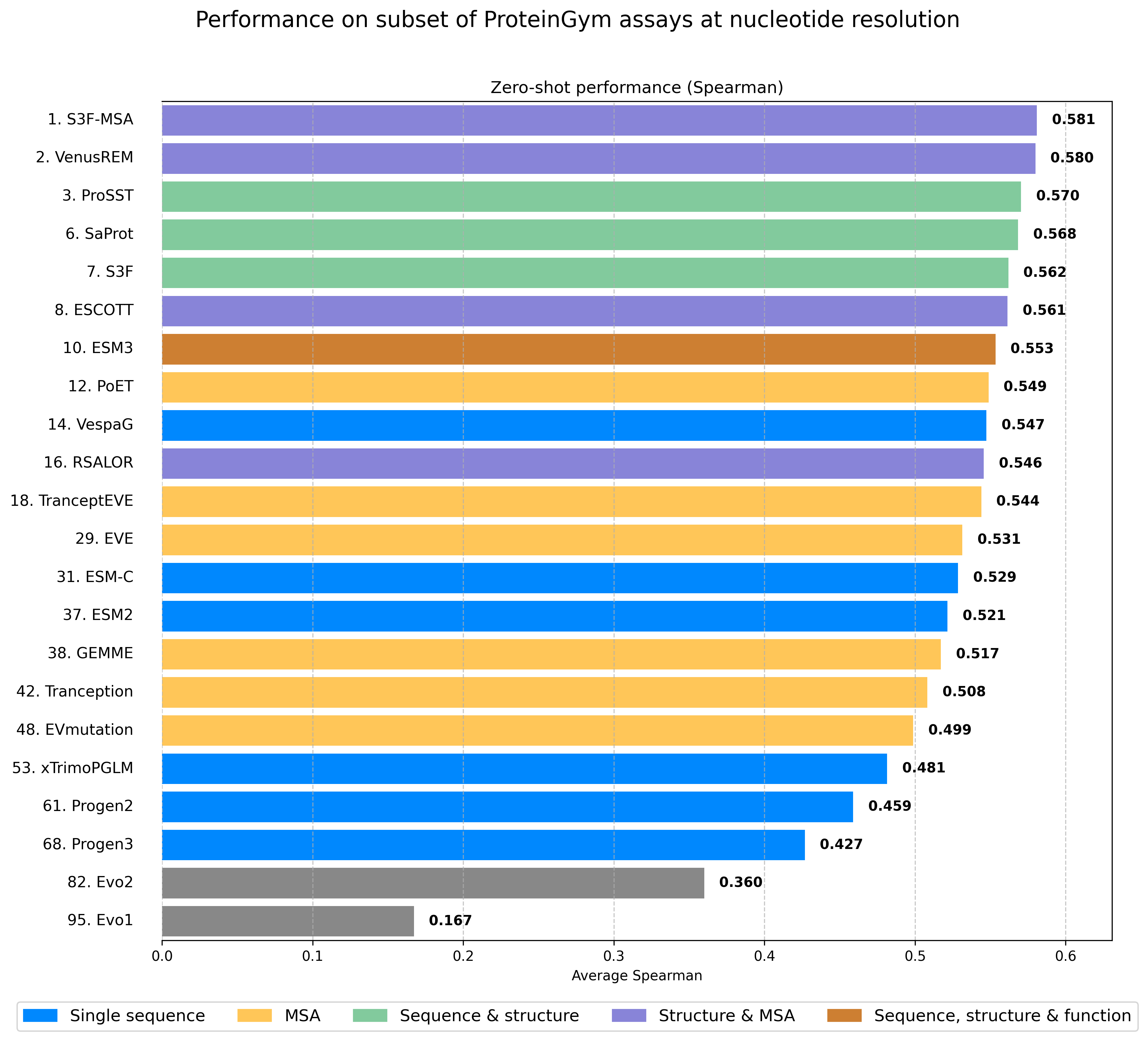
Figure 5 - ProteinGym Leaderboard on subset of assays mapped to nucleotide resolution. Average spearman across the 22 assays. Performance for Evo2 was obtained with the 7B checkpoint (which performs the best out of the various checkpoints).
Key findings:
Evo2 substantially outperforms its predecessor, Evo116
However, Evo2's performance remains far from SOTA—the best model on this subset (S3F-MSA) achieves a Spearman correlation of 0.582, while Evo2 reaches 0.360 and Evo1 only 0.167
Among the 97 baselines implemented in ProteinGym, Evo1 and Evo2 would rank #82 and #95 respectively
Notes:
Since Evo1 was trained exclusively on prokaryotes, this comparison might seem unfair. Restricting analysis to just the 11 prokaryotic assays, Evo1 achieves 0.300, Evo2 reaches 0.368, while the best protein-specific model (S3F-MSA) maintains a substantial lead at 0.578.
Evo2 performs very well on non-coding regions (a topic for another post!), but clearly lags behind protein-specific models for coding regions.
Where Do We Go From There?
1. Better retrieval strategies
In my view, improving sequence retrieval methods is the most important research direction to pursue in the short term. Given its significance, I'll expand on this topic in a dedicated follow-up post. For now, I'll highlight a few points directly related to the results discussed above.
Beyond the boost from retrieval for Tranception shown in Figure 4, you may have noticed RSALOR from Tsishyn et al. in the leaderboard from Figure 1. This approach makes zero-shot fitness predictions using only two components: a log-odds ratio (LOR) term derived from carefully regularized MSAs, and a RSA term that modulates the LOR based on residue surface accessibility (applying greater weight to core residues and less to surface positions). What's remarkable about this method is that it: a) incorporates both MSAs and structure, b) computes very simple metrics from these inputs, c) contains no trainable parameters, yet d) outperforms most protein language models (currently ranking #13 on both Spearman and NDCG benchmarks). This underscores a critical point: thoughtfully designed models using appropriate data modalities can outperform approaches relying on brute-force computational scaling. In this case, the bulk of the predictive signal comes from the MSA-based LOR term—highlighting the importance of effective retrieval approaches.
A parameter-free approach combining MSAs and structure currently outranks most billion-parameter language models—suggesting the most effective path forward may not be through larger models, but through smarter integration of evolutionary information and structural context.
Additionally, the MSAs used in ProteinGym could be further optimized. We previously demonstrated in the Tranception10 paper that crafting domain-specific MSAs substantially improves performance. Other studies have shown performance gains with alternative retrieval mechanisms; for instance, the ColabFold protocol outperforms the Jackhmmer protocol used in ProteinGym with certain model architectures (see Abakarova et al.18 or Truong Jr et al.19). More generally, approaches to craft the MSAs that are used to train or augment protein generative models have been mostly based on heuristics, sometimes developed for other tasks (eg., structure prediction), as opposed to being grounded in data and tailored for the task of interest.
2. Multimodal approaches leveraging improved structure predictions
As we saw above, the multimodal approaches integrating structure with MSAs that emerged in the past 12 months have driven significant performance improvements. Yet, the way that these modalities have been combined so far has been relatively rudimentary -- considerable room remains for enhancing how this combination is performed. Furthermore, similar to the point we made above about MSAs, the quality of the structures that are used in these multimodal approaches has a direct impact on downstream performance -- as structure prediction models improve, fitness prediction models will benefit. For instance, recent work from Sharma et al20 has demonstrated that for the subset of ProteinGym assays with available experimental structures in the PDB, using these structures rather than AlphaFold2-based predictions improved performance. Lastly, progress towards accurately predicting multiple conformers for a given sequence may also translate to further fitness prediction gains via ensembling.
3. Advanced post-training methods and alignment
This post has focused exclusively on zero-shot evaluation. In practice, leveraging even a small number of labels to predict properties of interest often yields substantial improvements. The recent Progen3 paper21 highlighted the benefits of scaling when combined with protein-specific alignment — ie. fine-tuning protein language models on limited experimental labels from a given protein family to better predict properties of unseen mutants for the same family.
This few-shot setting is highly relevant in practice, but fundamentally changes the discussion. It requires existing assays to collect sufficient labels, and developing these assays can be non-trivial depending on the protein and property of interest. What would be more useful in practice is protein-agnostic alignment, ie. aligning these pLMs with a handful of existing DMS assays and showing that it translates into performance gains for new proteins. It is fundamentally harder to pull off, but it would be a very strong case for scaling if it was helping there. This is a fascinating research direction as well, which will be covered in more detail in a future post.
Additional promising directions include synthetic data generation, hybrid ML+physics-based modeling architectures, and many others—but having already promised three additional blog posts above, I'll leave these topics for another day. Until next time!
Thanks
Special thanks to Daniel Ritter, Aaron Kollasch, Lood van Niekerk, Peter Groth, Artem Gazizov, Felix Teufel, Sebastian Prillo, Brian Schilder, Matsvei Tsishyn, Anton Bushuiev, Gianluca Lombardi, Yang Tan, Julius Schlensok, Timothy Truong Jr, and the other anonymous contributors for their help with PRs, PR reviews and the ProteinGym v1.3 code release in general. Thank you to the members of the Marks lab for helpful discussions. All errors are mine.
References
Tsuboyama et al, Mega-scale experimental analysis of protein folding stability in biology and design: https://www.nature.com/articles/s41586-023-06328-6
Beltran et al, Site-saturation mutagenesis of 500 human protein domains: https://www.nature.com/articles/s41586-024-08370-4
Open Datasets program from Align Bio foundation: https://alignbio.org/datasets-program
Feruz et al, Controllable protein design with language models: https://www.nature.com/articles/s42256-022-00499-z
Kaplan et al, Scaling Laws for Neural Language Models: https://arxiv.org/abs/2001.08361
Notin et al, ProteinGym: Large-Scale Benchmarks for Protein Fitness Prediction and Design: https://papers.nips.cc/paper_files/paper/2023/hash/cac723e5ff29f65e3fcbb0739ae91bee-Abstract-Datasets_and_Benchmarks.html
Fowler et al, Deep mutational scanning: a new style of protein science: https://www.nature.com/articles/nmeth.3027
Frazer et al, Disease variant prediction with deep generative models of evolutionary data: https://www.nature.com/articles/s41586-021-04043-8
Paul et al, Combining Structure and Sequence for Superior Fitness Prediction: https://openreview.net/forum?id=8PbTU4exnV
Notin et al, Tranception: Protein Fitness Prediction with Autoregressive Transformers and Inference-time Retrieval: https://proceedings.mlr.press/v162/notin22a.html
Gurev et al, Sequence-based protein models for the prediction of mutations across priority viruses: https://openreview.net/forum?id=DvC6VL7TJK
Hesselow et al, RITA: a Study on Scaling Up Generative Protein Sequence Models: https://arxiv.org/abs/2205.05789
Zhang et al., Protein language models learn evolutionary statistics of interacting sequence motifs: https://www.pnas.org/doi/10.1073/pnas.2406285121
Weinstein et al, Non-identifiability and the Blessings of Misspecification in Models of Molecular Fitness: https://doi.org/10.1101/2022.01.29.478324
Brixi et al, Genome modeling and design across all domains of life with Evo 2: https://www.biorxiv.org/content/10.1101/2025.02.18.638918v1
Nguyen et al, Sequence modeling and design from molecular to genome scale with Evo: https://www.science.org/doi/10.1126/science.ado9336
Tsishyn et al, Residue conservation and solvent accessibility are (almost) all you need for predicting mutational effects in proteins: https://www.biorxiv.org/content/10.1101/2025.02.03.636212v1
Abakarova et al, Alignment-based Protein Mutational Landscape Prediction: Doing More with Less: https://academic.oup.com/gbe/article/15/11/evad201/7344676
Truong Jr et al, PoET: A generative model of protein families as sequences-of-sequences: https://proceedings.neurips.cc/paper_files/paper/2023/hash/f4366126eba252699b280e8f93c0ab2f-Abstract-Conference.html
Sharma et al, Exploring zero-shot structure-based protein fitness prediction: https://arxiv.org/abs/2504.16886
Bhatnagar et al, Scaling unlocks broader generation and deeper functional understanding of proteins: https://www.biorxiv.org/content/10.1101/2025.04.15.649055v1
I tend to agree that pure scaling isn't the solution to PLMs. I wrote about this in a more narrative fashion about 2 years ago when the xTrimo model came out. I do suspect, however, that new techniques will be develop around scale, but not just parameter count.
https://biotechbio.substack.com/p/intuition-on-ai-scale-for-biologists
I thoroughly enjoyed reading this. It was inspiring that I always believe that scaling laws alone are not the ultimate solution for AI4Science. Besides, scientific tools should prioritize accessibility and usability, enabling private deployment and fine-tuning for specific research needs.
I noticed that you evaluated the performance of Evo2 on ProteinGym. You also mentioned that scoring Evo2 on ProteinGym requires mapping protein sequences back to their corresponding DNA sequences—a process you described as tedious and one that will eventually be extended to the full benchmark. May I kindly inquire if there is an estimated timeline for its release? Alternatively, would it be possible to share the 22 assays that have already been processed? We are eager to test the performance of our self-developed GLM, GENERator (https://arxiv.org/abs/2502.07272), on this benchmark.
For your interest (if there is any), GENERator is a generative GLM trained on the eukaryotic domain. Despite being 100 times cheaper and faster than Evo2, GENERator-3B has demonstrated competitive performance in Clinvar variant effect prediction (0.95 for GENERator vs. 0.96 for Evo2). If you'd like to explore further, we also provide a one-click VEP script for easy testing: https://github.com/GenerTeam/GENERator/blob/main/src/tasks/downstream/variant_effect_prediction.py.
On a related note, I wanted to share a minor comment regarding the suboptimal performance of Evo2. Based on my experience, part of this may be attributable to the limited length of CDS regions. For instance, augmenting the DNA sequence to 12k bp by including upstream non-coding regions could potentially improve performance. That said, I’m not entirely sure if this approach would be considered fair, as such additional information cannot be leveraged by any PLMs.